
Living Donor Liver Transplantation for Hepatorenal Fibroystic Diseaase in children
Rie Irie1, Atsuko Nakazawa2, Akinari Fukuda4, Seisuke Sakamoto4, Osamu Miyazaki3, Takako Yoshioka1, Mureo Kasahara4.
1Pathology, National Center for Child Health and Development, Tokyo, Japan; 2Clinical Research, Saitama Children's Medical Center, Saitama, Japan; 3Radiology, National Center for Child Health and Development, Tokyo, Japan; 4Organ Transplantation Center, National Center for Child Health and Development, Tokyo, Japan
Introduction: Hepatorenal fibrocystic disease, such as congenital hepatic fibrosis (CHF) and Caroli disease often accompany autosomal recessive polycystic kidney disease (ARPKD) which stems from a PKHD1 gene mutation. Liver transplantation (LT) is indicated for children with CHF who present liver cirrhosis or gastrointestinal bleeding due to portal hypertension, and children with Caroli disease who present recurrent cholangitis. The prognosis of these patients is thought to be good. In Japan, almost all LT donors to children are their parents, because cadaveric donor transplantation is not popular. However, parents of patients with hepatorenal fibrocystic disease are at risk of being heterozygote carriers of PKHD1. The aim of this study was to clarify the prognosis of those who received a living donor liver plantation (LDLT) from donors who might be heterozygote carriers of hepatorenal fibrocystic disease.
Patients and Methods: Fifteen patients with CHF and nine patients with Caroli disease underwent LDLT at National Center for Child Health and Development. The median age of the recipients was 7 years and 5 months (range; 1y1m to 14y3m). Fifteen and two patients were accompanied by ARPKD, and nephronophthisis, respectively. Ten patients underwent kidney transplantation. Sixteen of the donors were the recipients’ father, and nine donors were their mother. The median age of the donors was 37 years (range: 28y to 47y). The median follow-up period after transplantation was 4 years 5 months (range: 2m to 11y3m). We examined the histological and radiological findings of the donor livers and compared them with the findings of the recipients’ liver. We also examined complications in the recipients following the LDLT.
Results and Discussion: Eleven of the donor livers presented morphological abnormalities of the bile ducts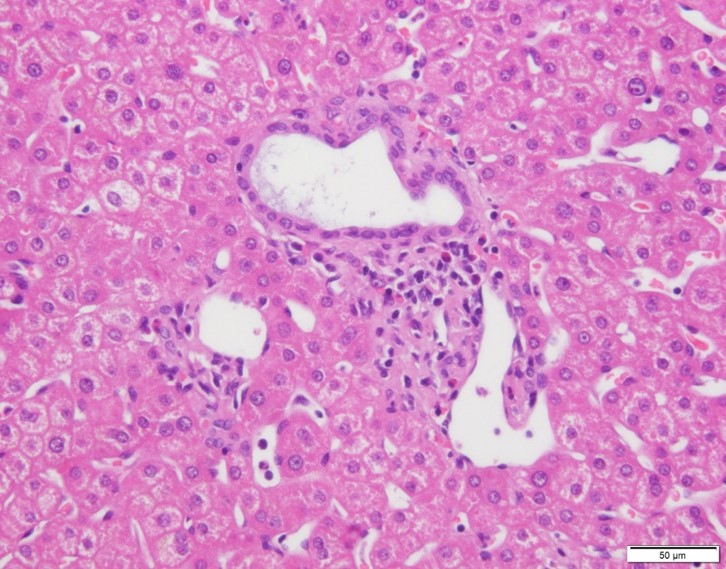 (three cases of dilated bile ducts, two cases of ductular reaction without inflammation, and six cases of small bile ducts). Liver cysts were found in eight donors by abdominal CT. Recipients’ complications included one death after LDLT due to infection and rejection. One patient underwent re-LT for graft failure due to antibody-mediated rejection and T-cell mediated rejection (TCMR). Two patients presented TCMR, one patient presented early chronic rejection, and two patients presented cholangitis. One patient presented dilated bile ducts, one patients presented a mild ductular reaction in the biopsy after LT, and one patient presented a dilatation of intrahepatic bile duct on abdominal CT six years after LT.
(three cases of dilated bile ducts, two cases of ductular reaction without inflammation, and six cases of small bile ducts). Liver cysts were found in eight donors by abdominal CT. Recipients’ complications included one death after LDLT due to infection and rejection. One patient underwent re-LT for graft failure due to antibody-mediated rejection and T-cell mediated rejection (TCMR). Two patients presented TCMR, one patient presented early chronic rejection, and two patients presented cholangitis. One patient presented dilated bile ducts, one patients presented a mild ductular reaction in the biopsy after LT, and one patient presented a dilatation of intrahepatic bile duct on abdominal CT six years after LT.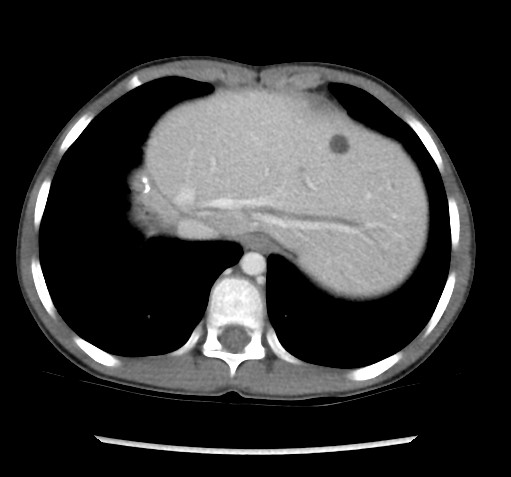 The overall survival rate was 96% (23/24), and the original graft survival rate was 92% (22/24).
The overall survival rate was 96% (23/24), and the original graft survival rate was 92% (22/24).
Conclusion: The prognosis of the recipients who received a LDLT from their parents for hepatorenal fibrocystic disease at our center was excellent. However, the histology of about half of the donor livers was abnormal. Careful follow-up is needed to insure long-time graft survival.

